- 58_[47类]轨迹分析和绘图.py
- 49_[扩展Beta]单质或化合物或多相混合体系密度计算.py
- 42_[扩展]化合物各类原子数统计.py
实现了从一个名为 "extract.xyz" 的文件中读取2列数据,基于这两列数据绘制了散点图和热力图
🎯 脚本功能
1. 计算多列数据表格中任意两列数据的线性相关系数(-1到1),并绘制热力图
2. 要求表格数据结构满足如下格式
Element Distribution X-O_BL X-Si_BL
P -1.277 1.57 2.33
Ti -0.1411 1.79 2.59
Mg 0.3604 1.99 2.82
计算公式:
基于ISAACS输出的径向分布函数g(r)文件,如g(r)[Ti,Si],计算 平均力势曲线,定义式如上所示,注意计算的是公式右边的部分。
数据处理过程中忽略了@开头的注释行,对于g(r)为0的部分采用nan进行填充,处理后的数据保存在pmf.txt文件中
1. 提示输入文件名,支持ISACAS导出的g(r)[Ti,Si]
2. 提示输入原子对的截断半径,需提前获取
- 源码:59_[指定x和y范围]自由能面二维投影图绘制.py
- 绘图的数据文件通常是
fes.dat文件,该数据文件仅包含3列数据,第1和2列数据 分别作为 x和y列,第三列数据是 z,3列数据分别使用空格或者tab分隔。
- 源码:58_[47类]轨迹分析和绘图.py
- 该脚本是
47_[类]轨迹分析和绘图.py升级版本
本脚本的功能如下:
01: 添加盒子信息到多帧xyz文件并输出各类原子范围。输入的xyz文件主要是基于VMD周期性处理过的,输出适用于plumed软件分析的xyz轨迹文件(依赖于其他方法)
02: 添加盒子信息,针对单帧/多帧xyz轨迹文件,且每一帧的原子数可以不相同(不依赖于其他方法)
03: 计算xyz轨迹文件某一帧的原子序号分布(不依赖于其他方法),主要是用于辅助plumed输入文件编写
04: 提取xyz轨迹文件某些范围帧数,如 1,3,5-10,30,每帧原子数可不同
05: cp2k输出的ener文件温度、势能随步数绘图,标准ener文件格式即可
06: 提取多帧xyz文件特定编号原子周围半径r范围内的原子[未考虑周期性]
07: 对xyz轨迹文件进行周期性扩增
08: 提取多帧xyz文件特定编号原子周围半径r范围内的原子[考虑周期性],功能 06升级版
09: 计算总的径向分布函数TRDF
10: 返回xyz轨迹文件盒子周期性扩增前后某一帧的原子序号分布(依赖于07方法,需要每一帧中同类原子连续分布)
11: xyz文件某一帧转data文件,data文件可用于lammps经典分子动力学模拟
12: colvar数据文件等绘图(自选列数),标准colvar文件格式即可
13: 计算xyz文件所有帧中某个原子到某个平面的距离,平面由三个原子的序号确定
14: 基于不同原子对的截断半径Rij,提取多帧xyz文件特定编号原子周围半径Rij范围内的配位原子[考虑周期性],功能 08升级版,适用于中心原子与配位原子是同种原子的情况,如B-B
15: 基于Sigmoid函数和不同原子对的截断半径Rij,计算多帧xyz文件特定编号原子的配位数[考虑周期性],基于功能 14 改编
16: 基于分段函数和不同原子对的截断半径Rij,计算多帧xyz文件特定编号原子的配位数[考虑周期性],基于功能 15 改编
17: 基于不同原子对的截断半径Rij,提取多帧xyz文件特定编号原子周围半径Rij范围内的配位原子[考虑周期性],基于功能 14 算法优化,适用于大体系快速计算,不显示盒子外配位补齐原子
18: 基于不同原子对的截断半径Rij,提取多帧xyz文件特定编号原子周围半径Rij范围内的配位原子[考虑周期性],基于功能 17 算法优化,适用于大体系快速计算,显示盒子外配位补齐原子
19: 提取某一帧某些编号的原子坐标,生成特定局域结构或团簇的原子坐标xyz文件
20: 基于不同原子对的截断半径Rij,针对各帧原子数不同的多帧xyz文件,提取某些类型中心原子的配位原子,基于方法18升级,不考虑周期性,适用于团簇结构的精细提取
-1: 测试
注意:
01: 该脚本的06,08,14功能可用于提取xyz轨迹文件中某个原子的轨迹(将截断半径设置为趋近于0的值的一个值,使得该截断半径内不出现配位原子),得到类似如下的多帧xyz轨迹文件,行数以3为周期,搭配 54_多帧轨迹文件中特定原子的xyz分坐标提取.py 脚本即可提取每一帧中的xyz分坐标。
1
This is number:1
B 0.303598 4.846121 6.626125
1
This is number:2
B 0.345071 4.648057 6.391401
1
This is number:3
B 0.351086 4.235202 6.350704
...
-
关于方法06、08、14、17和18的主要区别:
方法06:未考虑周期性,提取中心原子为中心,r为球形半径内的所有原子。方法08:考虑周期性,提取中心原子为中心,r为球形半径内的所有原子。方法14:考虑周期性,对不同原子对设置不同截断半径。使用周期性扩增,实际计算的原子数是输入体系的27倍,计算时间长,内存占用高。在每一帧中,部分配位原子可能被重复写入,但不影响结构可视化的正确性。方法17:考虑周期性,对不同原子对设置不同截断半径。优化算法,计算模拟盒子内所有满足符合要求的中心原子和配位原子。盒子边界处局域结构中的配位原子可能会显示不全,部分边界处配位原子远离局域结构,即使考虑周期性时该配位原子满足要求。方法18:在方法17基础上,对每一个配位原子相对中心原子的xyz分坐标进行周期性变换,使得计算时间短(1万原子体系计算时间约20分钟)、内存占用低。每一帧中配位原子不会被重复写入,边界处的局域结构显示完全,无满足配位要求但游离的配位原子。
-
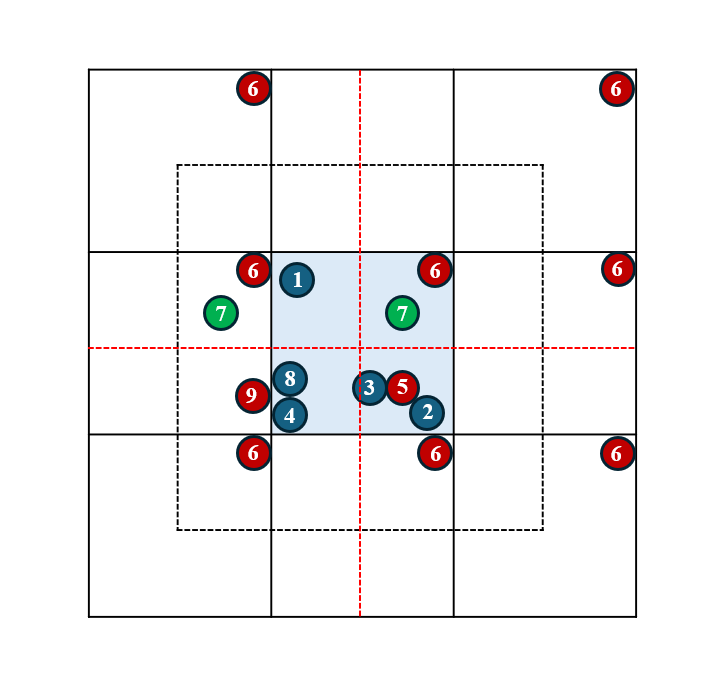
方法17和方法18局域配位结构算法分析- 代码核心思路:将所有帧中的所有原子(非扩增后盒子的原始原子,减少计算和内存占用)都写入到字典中,最外层for循环是帧数循环,然后次外层是每一帧中的所有j原子循环。接下来需要判断循环的原子类型(
中心i原子?非配位原子?配位原子j?)。最里面的一层循环是针对每一个中心原子i,每一帧中的每一个配位原子j是否满足截断半径距离要求(距离计算需要根据周期性条件进行变换)。如果满足要求,方法17会写入该j配位原子的原始真实坐标,且只会写入一次,无论该j配位原子是否满足多个中心原子的截断半径要求;方法18可能会在多个中心原子对j配位原子的距离循环判断中,写入多个j原子的坐标,且这些坐标都不同。 - 假如红色原子是配位原子,蓝色原子是中心原子。在方法14中,配位原子5(相对于中心原子2和3)和配位原子9(相对于中心原子8和4)都会在同一帧中被重复写入。例如,配位原子5相对于中心原子2会写入一次,相对于中心原子3还会写入一次,且两次写入的原子坐标均相同。
- 在
方法17中,配位原子9、配位原子5都只会被写入一次,9可能会显示为游离配位原子(远离中心原子8或4),这是因为原子9的坐标在写入时没有根据8或4的相对位置进行调整;中心原子8或者4的局域配位结构中可能会缺失配位原子9。 - 在
方法18中,配位原子6相对于中心原子1和2会分别调整分坐标,使其可视化时分别靠近相应的中心原子,以便补全结构。因此,配位原子6可能会被写入多次(分布在盒子外、分坐标不同),以便补全相应的局域结构。 - 中心原子1和配位原子6的距离计算:如果分坐标差值小于盒子边长一半,则使用该值;如果分坐标差值大于盒子边长一半,则为盒子边长减去该分坐标差值。
- 为了避免可视化时出现局域结构中配位原子缺失的问题,需要计算每一帧中配位原子相对于各个中心原子的相对分坐标。具体实现可参考
方法18核心代码。
- 代码核心思路:将所有帧中的所有原子(非扩增后盒子的原始原子,减少计算和内存占用)都写入到字典中,最外层for循环是帧数循环,然后次外层是每一帧中的所有j原子循环。接下来需要判断循环的原子类型(
⭐ 基于方法18和20提取xyz团簇结构文件:
-
方法20与方法18的区别方法20适用于各帧原子数不同的体系(原子编号不固定),不考虑周期性,不需要输入中心原子的编号,只需要输入中心原子种类即可,主要适用于团簇精细结构的提取。方法20处理的文件通常来源于方法18生成的xyz轨迹文件,包含满足指定原子对截断半径要求的局域结构或者粗糙团簇结构文件(使用原子对第一截断半径和第二截断半径都可以)。方法18考虑周期性,需要输入中心原子编号,各帧原子编号相对固定,各帧原子数一样。方法18基于原子对第二截断半径生成的文件后续可作为方法20的输入文件。
-
团簇结构提取思路1:
- 先使用
方法18基于原子对截断半径B O 4.5 B Si 3.4 B B 3.18 B Al 3.5提取某个中心原子(如B原子)的多帧xyz文件,注意B-O原子对截断半径是RDF的第二个谷值,对应B-O第二截断半径(该值可以稍微取大一些,不影响精细结构的准确性),其余B-Si,B-B,B-Al都是第一截断半径。获取的多帧xyz轨迹文件,是某个中心原子对应的粗糙团簇结构文件。该粗糙团簇最外层的部分O原子是多余的,但是其余原子都是准确的。 - 再使用
方法20基于原子对第一截断半径B O 2.2 Si O 2.2 Al O 2.5提取精细团簇结构(主要是各阳离子与O的第一截断半径,通常包括 X-O,Si-O等),去掉多余的O,B,Si,Al在提取过程中,不会发生删减。获取的多帧xyz轨迹文件是某个中心原子对应的精细团簇结构。 - 通过ovito、Jmol、VMD等可视化上述精细xyz轨迹文件,确定目标团簇结构对应的帧数,使用
方法04提取。
- 先使用
-
团簇结构提取思路2:
- 先用
方法18基于第二截断半径B O 4.5 B Si 3.4 B B 3.18 B Al 3.5,提取所有帧、所有中心原子对应的粗糙团簇结构。 - 然后使用
方法20基于第一截断半径B O 2.2 Si O 2.2 Al O 2.5提取所有帧、所有中心原子对应的精细团簇结构。 - 然后使用
Jmol可视化精细结构,找到目标团簇结构所在的帧数和中心原子编号(一个或多个中心原子编号),再使用方法04提取该帧xyz文件。 - 再使用
方法18基于某个中心原子编号和第二截断半径B O 4.5 B Si 3.4 B B 3.18 B Al 3.5,读取该帧xyz文件,提取该中心原子对应的精细团簇结构。 - 这种方法不需要记录某个精细团簇结构中所有中心原子和配位原子的编号,效率较高。
- 先用
⭐ 方法09注意事项:
-
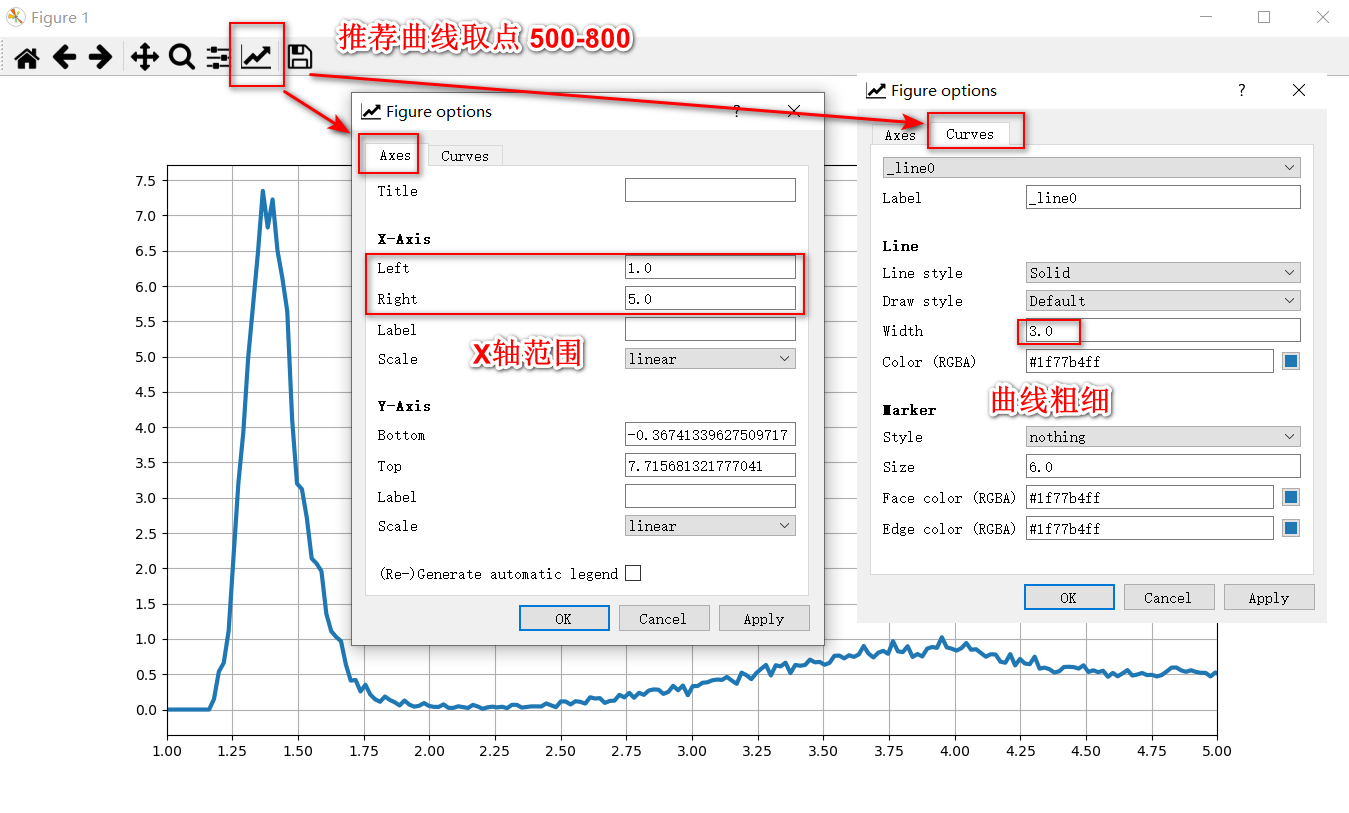
绘制rdf曲线默认取点数是500,推荐取点800,取点数太少会导致曲线的细节丢失,取点数太多会导致曲线有很多毛刺。
-
方法09代码中默认的x轴范围是[0,6],即只会计算该范围内的曲线,如果需要可以调整该范围。
# nPlotFrom = math.ceil(1.5/xdelta) # 1.5是指画图
nPlotFrom = 0
nPlotTo = math.ceil(6.0/xdelta)09方法绘制的曲线如下所示,可以在GUI窗口中设置X轴的范围,曲线的粗细等。
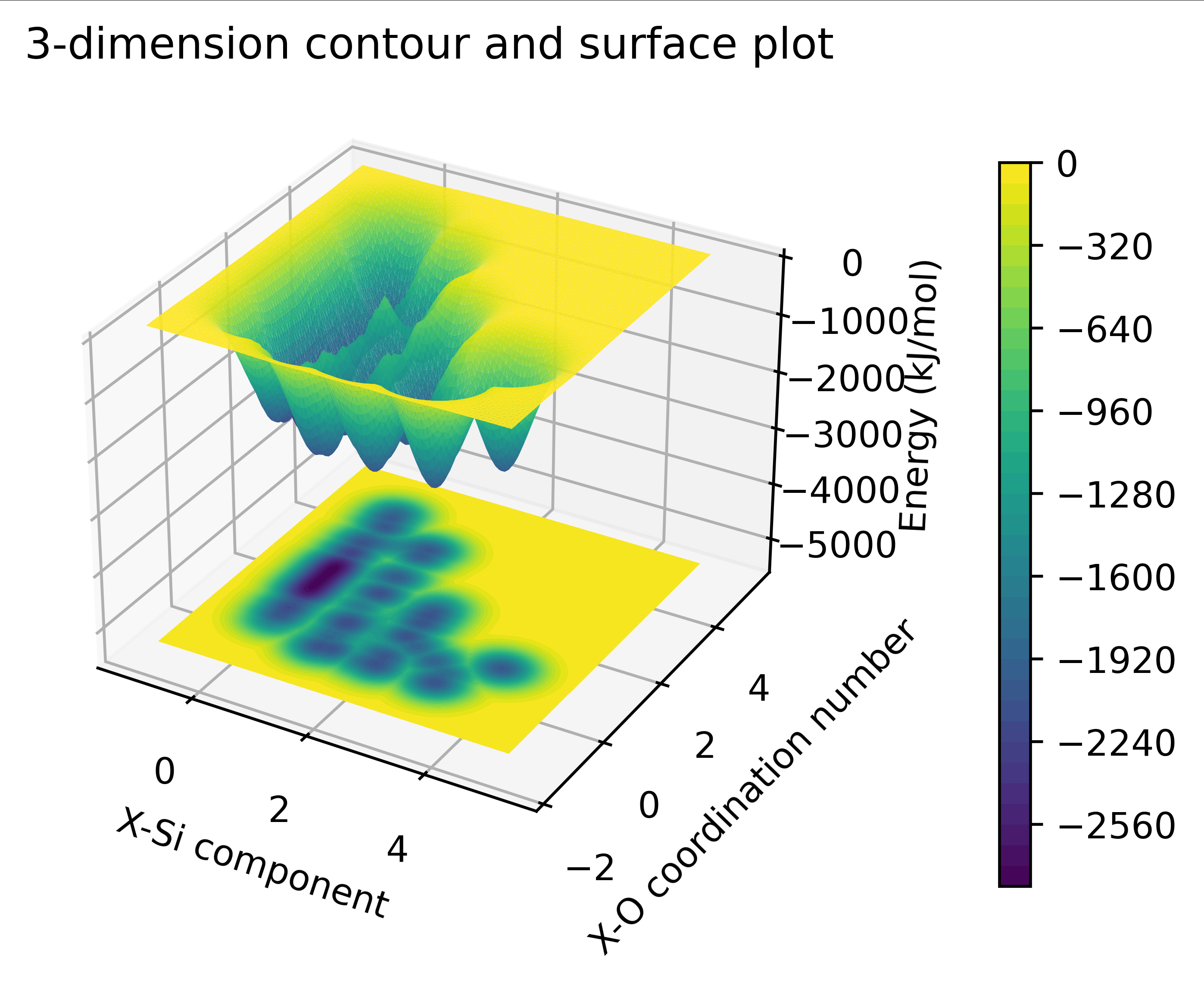
- 功能:绘制自由能的曲面及其二维填色图,需要初始化代码中的参数
1. 使用`np.loadtxt`加载名为`editColvar.txt`的数据文件。
2. 数据文件中的第一、二列分别代表x和y轴的数据,第三列是z轴的数据,z轴数据进行了单位转换以适应能量的表达(转换为kJ/mol)。
3. 设定了x、y、z轴的数据范围,并通过逻辑运算筛选出符合这些范围的数据点。
4. 定义了x和y的网格,通过griddata对原始散点数据进行插值,生成z轴的网格数据。
5. 在z轴某个固定的位置绘制等高线图(填色图),显示数据的等值线。
6. 颜色条的添加:为图形添加了颜色条,帮助解读不同颜色所代表的数据值。
- 示例
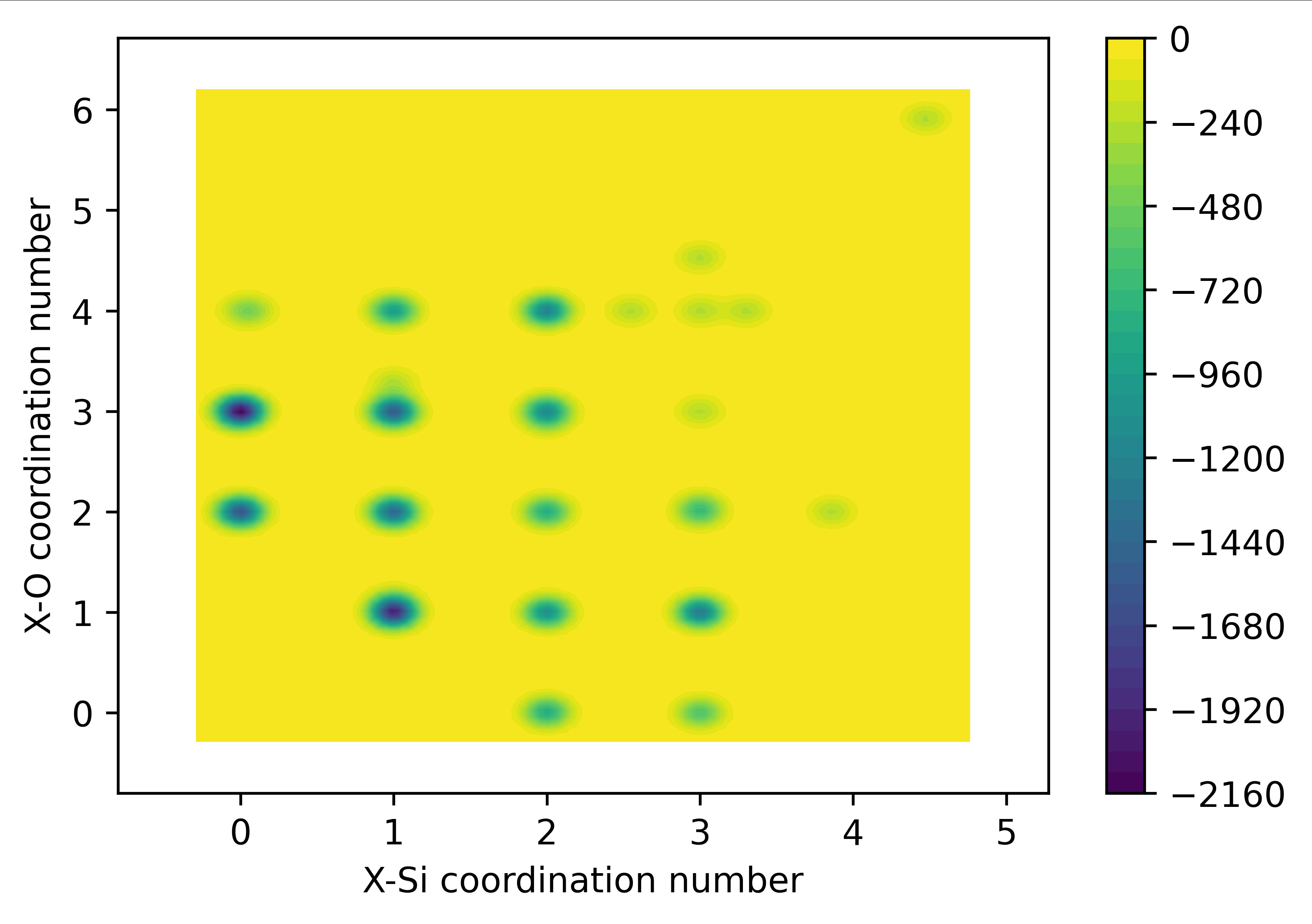
该脚本实现对 金属熔体中 特定杂质原子,如 X 原子周围配位的基体原子数量即占比的分析,基于百分比权重能计算平均配位数
这段代码的功能是:
获取当前目录下的所有文件并打印文件名。
提示用户输入待处理的数据文本名。
读取数据文本中的有效数据,并打印所有列的数据。
按照第一列的数值进行递增排序,并打印第一列及其对应的倒数第二列数据。
计算每一行中第一列和倒数第二列乘积的求和,并打印结果。
这段代码主要实现了文件操作、数据处理和统计计算的功能。
注意:该脚本在计算过程中忽略空行和#开头的行,并打印每一行中的第一列和倒数第二列乘积的求和(平均配位数),处理的数据参考格式如下
-------------------------------------------------------
# Environments for Mg atoms:
# N(tot) N(Mg) N(Si) Number or Percent
10 0 10 1.30464 or 16.308 %
11 0 11 1.71523 or 21.440 %
13 0 13 1.42384 or 17.798 %
12 0 12 1.90728 or 23.841 %
14 0 14 0.72185 or 9.023 %
9 0 9 0.53642 or 6.705 %
15 0 15 0.16556 or 2.070 %
16 0 16 0.04636 or 0.579 %
8 0 8 0.14570 or 1.821 %
7 0 7 0.01325 or 0.166 %
6 0 6 0.00662 or 0.083 %
17 0 17 0.01325 or 0.166 %
- 从
fes.dat文件中读取三维坐标数据(x, y, z),并在二维网格上找到并可视化每个网格内的最小z值 - 基于该图,通过对比gnuplot绘制的二维填色图,可以确定势能面上各个局域极小值的xyz分坐标
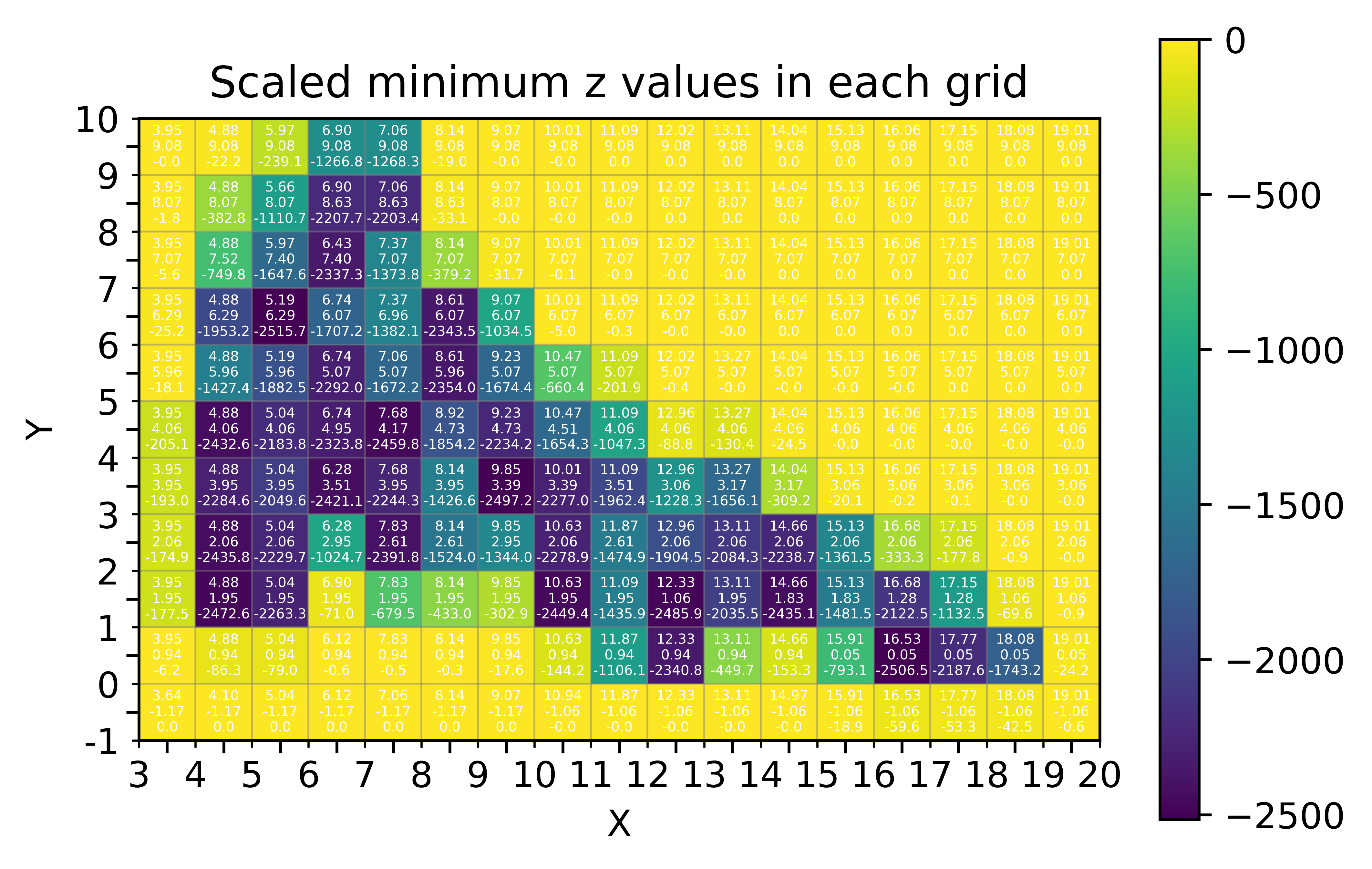
相比于 55_plot_min_z_grid_values.py,热图单元格中的数据标签与x和y轴的刻度是正确对应的
- 注意该脚本绘制的热图单元格中的数据标签与x轴和y轴上的刻度不一致,需要优化
- 基于该图,通过对比gnuplot绘制的二维填色图,可以确定势能面上各个局域极小值的xyz分坐标
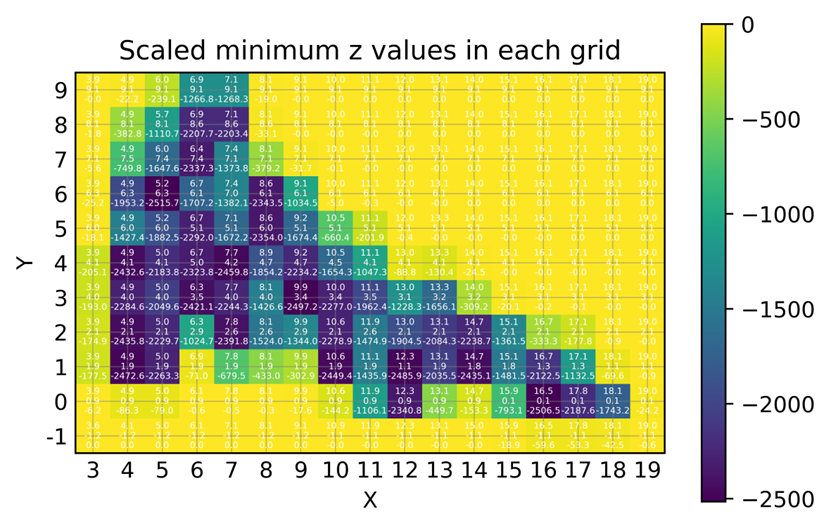
- 通过gnuplot大概判断局域极小值的x和y取值范围
- 在
55_plot_min_z_grid_values.py脚本中初始化如下代码参数,包括x和y的局域极小值的大概范围
find_min_z_and_corresponding_x_and_y(4, 6, 3, 5, 'fes.dat')
- 如下所示为显示示例,打印z的局域极小值
Minimum z value for x in range [4, 6] and y in range [3, 5]: -0.9265716133
Corresponding x value: 4.881
Corresponding y value: 4.063
Product of minimum z with 4.3597*6.022*100: -2432.632
本脚本的功能如下:
01: 提取每一帧只有1个原子的xyz轨迹文件中该原子的x或y或z分坐标,输出文件有两列,第一列为帧数,第二列为分坐标
注意:单原子轨迹文件可基于本文件夹下47或58脚本获取
01: 导入必要的模块:使用import语句导入numpy和matplotlib.pyplot模块。
02: 定义常量和变量:设置一些常量和变量,如长度单位转换因子、玻尔到埃单位转换因子、玻尔兹曼常数等。
03: 加载colvar文件:使用np.loadtxt()函数加载指定路径下的colvar文件,并将数据存储在colvar_raw变量中。
04: 提取CVs数据:从colvar_raw中提取两个关键变量(CVs)的数据,分别存储在d1和d2变量中。
05: 创建二维直方图:使用np.histogram2d()函数根据d1和d2的数据生成二维直方图,将结果存储在cv_hist变量中。
06: 打印直方图的bin边缘和频率:通过打印cv_hist的元素来显示直方图的bin边缘和频率信息。
07: 计算概率:将直方图中的频率归一化,得到每个bin的概率,存储在prob变量中。
08: 计算自由能表面:根据概率值,使用玻尔兹曼常数和温度计算自由能表面,并将结果存储在fes变量中。
09: 转换能量单位为kJ/mol:将自由能表面的能量单位从eV转换为kJ/mol,得到fes_kjmol变量。
10: 绘制并保存自由能表面图像:根据计算得到的自由能表面数据,使用plt.imshow()函数绘制图像,并使用plt.colorbar()添加颜色条。然后设置坐标轴标签和图像标题,并使用plt.show()显示图像。注释掉的代码部分是用于保存图像的。
两相模型构建流程:
01: 确定单相/均相的总原子数及各组元(化合物)的质量百分数,基于脚本 49_[扩展Beta]单质或化合物或多相混合体系密度计算.py 计算各组元的摩尔数及体系的密度
02:基于结合42_[扩展]化合物各类原子数统计.py 计算体系中各类原子数量,利用material studio进行建模,获取.cell文件
03:结合ovito或脚本48_[类]cell文件转data文件.py 导出 xyz格式的初始结构模型,进行标准AIMD获取平衡结构,提取某两帧平衡结构model1.xyz和model2.xyz作为搭建两相结构的初始结构模型
04:利用本脚本功能1对上述其中一个model2.xyz模型的z分坐标加上一个常数(model1.xyz模型的晶格常数C + 两相界面真空层厚度0.3~0.5 Å),获取modify_model2.txt模型文件
05:利用本脚本02功能对上述 model1.xyz 和 modify_model2.txt 进行合并,得到两相模型 model_merge.xyz,注意添加两相模型总原子数以及晶格参数
06:利用本脚本03和04功能获取model_merge.xyz中z轴方向需要在边界处固定的原子编号,添加到cp2k的inp文件中
07:利用上述model_merge.xyz 进行无偏分子动力学模拟,获取两相模型的平衡结构 merge_equilibrium.xyz
08:在inp文件中设置偏置参数,利用上述merge_equilibrium.xyz模型进行元动力学模拟
本脚本的功能如下:
01: txt文件特定列加上常数,忽略 空行 和 # 开头行,打印总行数(总行数不计空行和 # 字开头行)
02: 将两个txt文件合并,忽略 空行 和 # 开头行,打印总行数(总行数不计空行和 # 字开头行)
03: 输出txt文件某列 大于 等于某个数的行号,行号包含#开头行
04: 输出txt文件某列 小于 等于某个数的行号,行号包含#开头行
05: 生成10000行自定义曲面函数的data.txt文件
06: 绘制3列(x,y,z)数据组成的曲面图及其二维填色图
-1: 测试
本脚本的功能如下:
01: 创建json势参数数据库文件,初始内容为 {"charge": {}, "parameter": {}}
02: 添加势函数原子电荷信息到数据库文件中
03: 添加势函数原子对势参数信息到数据库文件中
04: 查看势参数数据库内容,满足所有json格式数据库的查看
05: 输出控制lammps运行的in文件(目前主要为元素相互作用势参数部分)
-1: 测试
1. 运行示例
输入想要采用的原子顺序,需要与data文件中的一致,用英文逗号隔开,如: Si,B,Ca,O 网络形成体在前,然后是碱金属原子,最后是O原子
Si,Al,B,Ca,O
势参数数据库中不存在: Si-Si 或 Si-Si 原子对的势参数
势参数数据库中不存在: Si-Al 或 Al-Si 原子对的势参数
势参数数据库存在该原子对势参数 Si-B
势参数数据库中不存在: Si-Ca 或 Ca-Si 原子对的势参数
势参数数据库存在该原子对势参数 Si-O
势参数数据库中不存在: Al-Al 或 Al-Al 原子对的势参数
势参数数据库中不存在: Al-B 或 B-Al 原子对的势参数
势参数数据库中不存在: Al-Ca 或 Ca-Al 原子对的势参数
势参数数据库存在该原子对势参数 Al-O
势参数数据库存在该原子对势参数 B-B
势参数数据库中不存在: B-Ca 或 Ca-B 原子对的势参数
势参数数据库存在该原子对势参数 B-O
势参数数据库中不存在: Ca-Ca 或 Ca-Ca 原子对的势参数
势参数数据库存在该原子对势参数 Ca-O
势参数数据库存在该原子对势参数 O-O
set type 1 charge 1.89 # Si
set type 2 charge 1.4175 # Al
set type 3 charge 1.4175 # B
set type 4 charge 0.945 # Ca
set type 5 charge -0.945 # O
group Si type 1
group Al type 2
group B type 3
group Ca type 4
group O type 5
pair_coeff * * 0.00000000 1.000 0.00000000 # others
pair_coeff 1 3 337.70 0.29 0.0 # Si-B
pair_coeff 1 5 50306.10 0.161 46.2978 # Si-O
pair_coeff 2 5 28538.42 0.172 34.5778 # Al-O
pair_coeff 3 3 484.40 0.35 0.0 # B-B
pair_coeff 3 5 206941.81 0.124 35.0018 # B-O
pair_coeff 4 5 155667.70 0.178 42.2597 # Ca-O
pair_coeff 5 5 9022.79 0.265 85.0921 # O-O注意:
输入想要采用的原子顺序,需要与data文件中的一致,用英文逗号隔开,如: Si,B,Ca,O 网络形成体在前,然后是碱金属原子,最后是O原子,这是因为需要保证pair_coeff中第二列原子的编号要比第一列原子编号要大,否则会报错。- data文件中的原子顺序和编号可以通过VESTA等软件查看,也可以使用
58_[47类]轨迹分析和绘图.py脚本的功能03分析.xyz文件获取。
2. 势参数
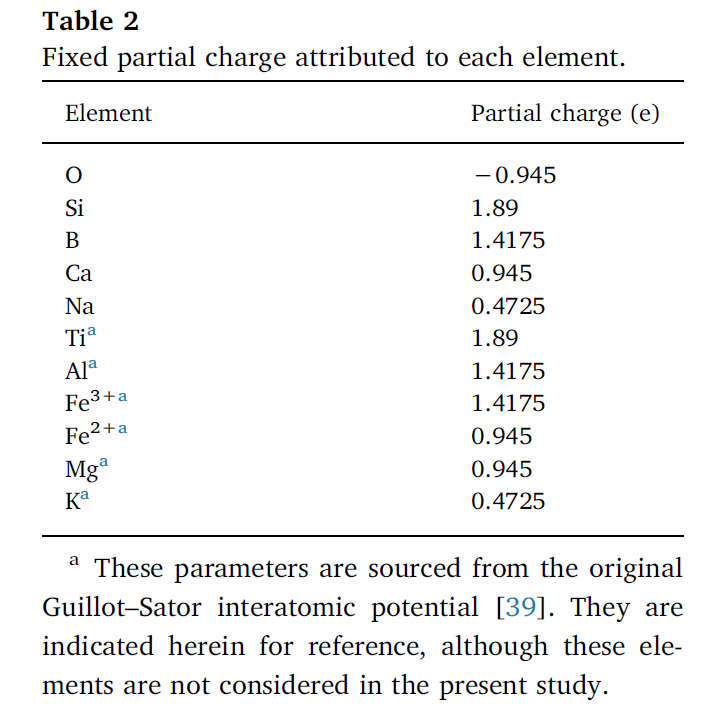
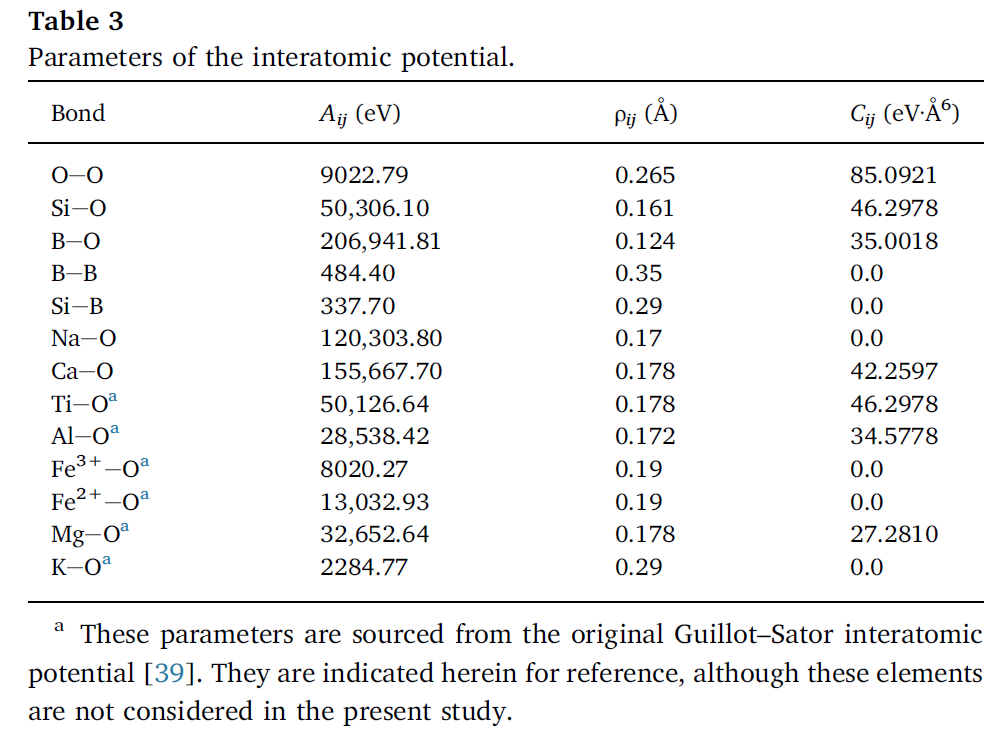
3. 参考资料
- Wang M, Krishnan N M A, Wang B, et al. A new transferable interatomic potential for molecular dynamics simulations of borosilicate glasses[J]. Journal of Non-Crystalline Solids, 2018, 498: 294-304.
❤️ 1. 新增功能
- 该脚本相比于
49_[扩展]单质或化合物或多相混合体系密度计算.py新增功能05: 基于各组分质量比计算各原子数量 - 整合
42_[扩展]化合物各类原子数统计.py脚本功能,能够同时计算混合体系(单质和化合物或纯化合物)中各类原子数量以及总原子的数量,便于 Materials studio建模(该功能耦合在功能06中)。 - 新版脚本位于
C:\Users\sun78\Desktop\cp2k_model\50_SiV\49_[扩展Beta]单质或化合物或多相混合体系密度计算.py目录下
⭐ 2. 脚本说明
本脚本的功能如下:
01: 查看数据库内容
02: 手动修改、添加或删除化合物原子组成数据库中的数据
03: 手动修改、添加或删除密度数据库中密度数据,并保存为新的数据库文件
04: 基于factsage体积数据计算单组元密度
05: 基于各组分质量比计算各原子数量
06: 计算多组元混合体系密度
-1: 测试
⭐ 3. 特色功能
- 该脚本既可以单独基于 1 mol 化合物体积(Factsage)计算其密度,也可以计算二元以及多元组分混合体系的密度(功能04和06)
- 该脚本能够将组元的摩尔组成转换为各类原子的数量,并显示各组元的摩尔比和质量百分比(功能06)
- 该脚本能够基于各组元的质量百分比计算摩尔比,可用于建模(功能05)。后续可以基于各组元摩尔数计算各类原子数以及密度,便于使用
Materials studio建模(功能06) - 如果组元质量百分数不固定,但是碱度一定时,可以参考
01_固定碱度下改变B2O3含量组分变化.py脚本计算各组元质量百分数

注意:可以在total中查看总原子数,如果运行结果中缺少上述数据,注意检查脚本是否为最新版本。
⭐ 4. 建模流程
- 参考materials project、GPT4、谷歌学术等,找到最稳定氧化物
-
确定均相或两相体系的组分以及摩尔数。如果已知体系中各组元的质量百分数,可以使用
49_[扩展Beta]单质或化合物或多相混合体系密度计算.py脚本功能 05 计算各组元的摩尔数。 -
向
49_[扩展-1化合物原子组成数据库]单质或化合物或多相混合体系密度计算.json数据库中添加稳定氧化物的组成,例如 PtO2- 利用 Factsage 的
View Data模块查找各氧化物组元的密度,并写入到49_[扩展-2化合物密度数据库]单质或化合物或多相混合体系密度计算.json数据库中。 - 对于文献中或者factsage的
View Data模块缺失的化合物密度,例如PtO2,可以尝试使用Reaction模块计算体积数据进行倒推(View Data模块缺密度的化合物似乎无法用于Reaction模块多元体系的体积和密度计算)。 - 注意:
Materials project上提供每种晶体结构的密度数据(Density column,按照Energy above hull从小到大排列)。
- 利用 Factsage 的
-
使用脚本
49_[扩展Beta]单质或化合物或多相混合体系密度计算.py功能 06 获取体系密度,同步获得体系中各类原子的数量以及总数,便于 Materials Studio 建模。 -
Materials Studio 建模,注意将建模的盒子边长等信息粘贴到ppt中,后期需要添加到
.xyz文件中 -
编写 cp2k inp控制文件,进行分子动力学模拟。
⭐ 5. 计算固定碱度下各组元质量百分比
- 背景
假设某个体系中含有CaO,SiO2,Al2O3,B2O3四种组元,且碱度C/S是定值,当改变B2O3含量时,计算各组元的质量百分数。
-
代码思路
- 提示用户输入
Al2O3质量分数a和B2O3质量分数b,单位是%,如10,1,使用英文逗号分隔 - 参数
k代表CaO和SiO2质量分数之比,设置参数k=0.6 - 计算SiO2质量分数y,计算公式为
y=(100-a-b)/(k+1) - 计算CaO质量分数x,计算公式为
x=k*y - 输出x和y的值
- 提示用户输入
-
代码实现
01_固定碱度下改变B2O3含量组分变化.py
# 提示用户输入Al2O3和B2O3的质量分数
input_str = input("请输入Al2O3质量分数a和B2O3质量分数b,单位是%,如10,1,使用英文逗号分隔:")
a_str, b_str = input_str.split(",") # 拆分输入的字符串
a = float(a_str) # 转换为浮点数
b = float(b_str) # 转换为浮点数
# 提示用户输入碱度C/S参数k
k_str = input("请输入碱度C/S参数k:")
k = float(k_str) # 转换为浮点数
# 计算SiO2的质量分数y
y = (100 - a - b) / (k + 1)
# 计算CaO的质量分数x
x = k * y
# 输出计算结果
print(f"CaO的质量分数 x = {x:.2f}%")
print(f"SiO2的质量分数 y = {y:.2f}%")⭐ 该脚本是 38_单质或化合物或多相混合体系密度计算.py 升级版本
本脚本的功能如下:
01: 查看数据库内容
02: 手动修改、添加或删除化合物原子组成数据库中的数据
03: 手动修改、添加或删除密度数据库中密度数据,并保存为新的数据库文件
04: 基于factsage体积数据计算单组元密度
05: 计算多组元混合体系密度
-1: 测试
49_[扩展-1化合物原子组成数据库]单质或化合物或多相混合体系密度计算.json
化合物原子组成数据库,对应上述脚本功能2
{"CaO": {"Ca": "1", "O": "1", "total": "2"}, "SiO2": {"Si": "1", "O": "2", "total": "3"}, "B2O3": {"B": "2", "O": "3", "total": "5"}, "P2O5": {"P": "2", "O": "5", "total": "7"}, "MgO": {"Mg": "1", "O": "1", "total": 2}, "Al2O3": {"Al": "2", "O": "3", "total": 5}, "K2O": {"K": "2", "O": "1", "total": 3}, "TiO2": {"Ti": "1", "O": "2", "total": 3}, "V2O5": {"V": "2", "O": "5", "total": 7}, "Cr2O3": {"Cr": "2", "O": "3", "total": 5}, "MnO": {"Mn": "1", "O": "1", "total": 2}, "MnO2": {"Mn": "1", "O": "2", "total": 3}, "FeO": {"Fe": "1", "O": "1", "total": 2}, "Fe3O4": {"Fe": "3", "O": "4", "total": 7}, "Fe2O3": {"Fe": "2", "O": "3", "total": 5}, "Co2O3": {"Co": "2", "O": "3", "total": 5}, "NiO": {"Ni": "1", "O": "1", "total": 2}, "Cu2O": {"Cu": "2", "O": "1", "total": 3}, "CuO": {"Cu": "1", "O": "1", "total": 2}, "ZrO2": {"Zr": "1", "O": "2", "total": 3}, "CaF2": {"Ca": "1", "F": "2", "total": 3}, "BaO": {"Ba": "1", "O": "1", "total": 2}, "La2O3": {"La": "2", "O": "3", "total": 5}, "Ce2O3": {"Ce": "2", "O": "3", "total": 5}, "MoO2": {"Mo": "1", "O": "2", "total": 3}}49_[扩展-2化合物密度数据库]单质或化合物或多相混合体系密度计算.json
化合物密度数据库,对应上述脚本功能3,这些氧化物的密度数据有些是从书本中查询得到的,有些是利用factsage计算得到的
{"SiO2": ["2.335", "g/cm3", "null", "SiO2", "null", "null", "null"], "CaO": ["2.8581", "g/cm3", "null", "CaO", "null", "null", "null"], "B2O3": ["2.55", "g/cm3", "null", "B2O3", "null", "null", "null"], "V2O5": ["3.357", "g/cm3", "null", "V2O5", "null", "null", "null"], "MnO2": ["5.2", "g/cm3", "null", "MnO2", "null", "null", "null"], "Fe2O3": ["5.277", "g/cm3", "null", "Fe2O3", "null", "null", "null"], "TiO2": [4.245019666206016, "g/cm3", "null", "TiO2", "null", "null", "null"], "Al2O3": [3.987067500879834, "g/cm3", "null", "Al2O3", "null", "null", "null"]} 本脚本的功能如下:
01: Material Studio分数坐标cell文件转 Multiwfn标准格式的xyz笛卡尔坐标文件
02: Material Studio分数坐标cell文件转 lammps标准格式的data结构文件
-1: 测试
注意:运行上述脚本功能02会提示输入原子顺序,以便生成的data文件中按照该顺序排列原子种类。新增支持输出 atomic 原子格式的 lammps data 文件,在功能2运行过程中会提示选择 charge 格式还是 atomic 格式,atomic 格式是deepmd运行lammps支持的标准格式。
本脚本的功能如下:
01: 添加盒子信息到多帧xyz文件并输出各类原子范围。输入的xyz文件主要是基于VMD周期性处理过的,输出适用于plumed软件分析的xyz轨迹文件(依赖于其他方法)
02: 添加盒子信息,针对单帧/多帧xyz轨迹文件,且每一帧的原子数可以不相同(不依赖于其他方法)
03: 计算xyz轨迹文件某一帧的原子序号分布(不依赖于其他方法),主要是用于辅助plumed输入文件编写
04: 提取xyz轨迹文件某些范围帧数,如 1,3,5-10,30,每帧原子数可不同
05: cp2k输出的ener文件温度、势能随步数绘图
06: 提取多帧xyz文件特定编号原子周围半径r范围内的原子[未考虑周期性]
07: 对xyz轨迹文件进行周期性扩增
08: 提取多帧xyz文件特定编号原子周围半径r范围内的原子[考虑周期性]
09: 计算总的径向分布函数TRDF
10: 返回xyz轨迹文件盒子周期性扩增前后某一帧的原子序号分布(依赖于07方法,需要每一帧中同类原子连续分布)
11: xyz文件某一帧转data文件,data文件可用于lammps经典分子动力学模拟
-1: 测试
''')
print("请选择功能,输入Enter默认为-1测试") # 提示选择功能
该脚本是 42_化合物各类原子数统计.py 升级版本
⭐ 目前该脚本功能已经整合到49_[扩展Beta]单质或化合物或多相混合体系密度计算.py脚本中,功能6(计算混合体系密度)可直接计算体系中各类原子数
请输入待处理字典,或字符串,如 {'CaO': '11', 'SiO2': '10', 'B2O3': '1'} 或 str,CaO,11,SiO2,10,B2O3,1 ,其中开头的 str 必不可少
代码实现了一个化学计算器,可以根据输入的化学式和化合物的数量,计算出凝聚态体系中各种化合物的原子组成。具体实现过程如下:
1. chemicalsSplit()函数将输入的化学式字符串转换为字典,字典的键为分子式,值为该分子式的原子组成。
2. loadData()函数从json文件中读取数据,返回一个字典,该字典包含了各种分子式的原子组成。
3. 判断输入的分子式的组成是否都在数据库中,如果有缺失的分子式,则调用chemicalsSplit()函数,为其添加原子组成,并将更新后的字典写入到json文件中。
4. atomCount(atomDict,compoundDict)函数根据输入的分子式的组成字典和原子组成数据库,返回一个列表,列表的元素是字典,每个字典是每种化合物的各原子构成。
5. dict_Sum(ini_dict)函数将列表中多个字典进行合并,键相同的值相加。
综上所述,该代码实现了一个化学计算器,可以方便地计算出凝聚态体系中各种化合物的原子组成。
请输入包含有中心原子和配位原子的字典,如 {'1': ['16', '27'], '2': ['12']} 格式,该字典可以基于脚本40计算出来
推荐使用 脚本40 进行计算
1. txt中的数据格式参考 40_键级密度百分比.py 注释中列出的键级数据,每列数据中可能就会有"--"字符串,不同列之间使用tab进行分隔
2. 使用dataframe数据结构读取该数据文件,然后打印出每列数据的最大值和最小值
📌 脚本说明
1. 数据来源与 [40_键级密度百分比.py] 脚本相同
2. 统计各偶数列的的数据个数,即模拟体系中各类化学键的数量,计算各类化学键的百分比
3. 可以直接将运行结果中的最后数据列部分复制到origin中绘制饼图
🎯 结果展示
请输入要处理的键级txt文件名: Ti-O.txt
列2中化学键的个数:158, '--'的个数:133
列4中化学键的个数:291, '--'的个数:0
列6中化学键的个数:38, '--'的个数:253
每一列的总行数:291
总的化学键数量为: 487.0
列2中化学键的百分比:32.44 %
列4中化学键的百分比:59.75 %
列6中化学键的百分比:7.8 %
32.44
59.75
7.8
📌 数据来源
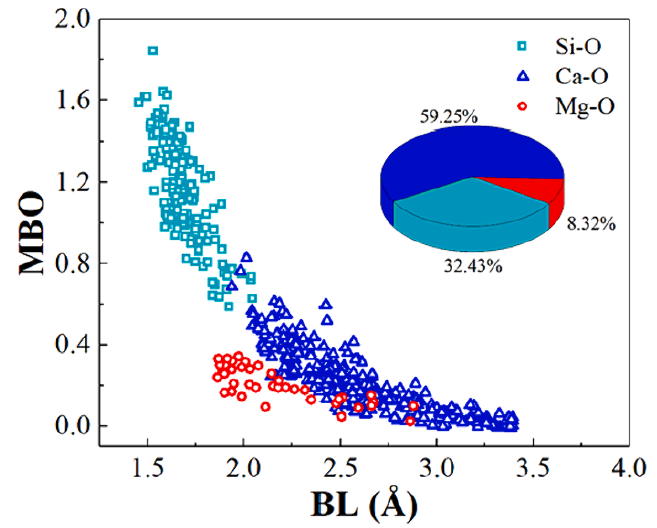
如下所示是一个体系中不同原子对的键长-键级分布数据,奇数列是键长数据,偶数列是键级数据
由于同一体系中不同类型化学键的数量不同,因此采用'--'符号填补缺失的数据,使得所有列的数据个数一致
下面的数据通常是从origin中复制粘贴过来的,origin中可以使用'--'符号填补空白
1.654 0.983 3.382 0.033 1.76 1.656
1.656 1.493 2.936 0.04 1.756 1.028
1.566 1.075 3.137 0.036 1.887 0.722
1.612 1.318 2.324 0.282 1.813 1.105
1.728 0.956 2.835 0.145 2.093 0.783
1.664 1.472 2.151 0.467 1.766 1.732
1.766 0.929 2.492 0.115 2.09 0.574
1.572 1.037 2.855 0.131 -- --
1.791 0.842 2.375 0.286 -- --
1.707 1.234 2.399 0.115 -- --
1.644 1.278 2.023 0.53 -- --
1.552 1.419 2.377 0.14 -- --
1.699 0.982 2.254 0.309 -- --
1.717 0.967 2.299 0.371 -- --
-- -- 3.1 0.039 -- --
-- -- 2.363 0.259 -- --
-- -- 2.174 0.384 -- --
-- -- 2.649 0.122 -- --
-- -- 2.462 0.299 -- --
-- -- 2.775 0.088 -- --
-- -- 3.016 0.008 -- --
-- -- 2.663 0.093 -- --
-- -- 3.18 0.022 -- --
🔥 基本概念
总键级(TBO):体系中各类化学键的键级数据求和。
总键级密度(TBOD):TBO/V,总键级/模拟盒子体积,体积的长度单位通常采用 Å。
Partial bond order density(PBOD):各类键的键级总和/模拟盒子体积。
键级密度百分比:各类键的PBOD/TBOD*100% , 也等于 各类键的各类键的键级总和/总键级*100%
参考文献:https://doi.org/10.1063/1.4987033
💡 脚本说明
1. 这段代码的目的是处理包含键级数据的文本文件,计算偶数列的总和、总和占比和键序密度等信息,并将这些信息输出到控制台。
2. 由于需要计算键序密度,因此计算过程中需要输入模拟盒子的边长,默认采用立方体盒子。
🔔 结果展示
请输入要处理的键级txt文件名: Ti-O.txt
请输入电子结构计算的立方模型盒子边长,单位, Å:15.0
[184.48100000000002, 57.681000000000004, 35.66099999999999]
各偶数列的和: [184.48100000000002, 57.681000000000004, 35.66099999999999]
总和: 277.82300000000004
各偶数列的和占总和的百分比: [66.4, 20.76, 12.84]
体系中各化学键的键序密度: [0.0547, 0.0171, 0.0106]
总键序密度: 0.0823
- 源码1:40_同种原子键级计算.py
- 源码2:40_同种原子键级计算_2nd.py
1. 上述两个脚本的功能是一样的,用于清洗 [40_Mayer键级与键长_升级版.py] 脚本输出的同类原子对无效或重复的键级数据
2. 基于 [40_Mayer键级与键长_升级版.py] 脚本计算同类原子键级数据时,输出的 [bondOrder.txt] 文件中同类原子数据会有重复,
以SiP体系为例,假如有a个Si原子,b个P原子,计算Si-Si的键级,如果 [40_Mayer键级与键长_升级版.py] 输出的Si-Si键级数据有
c 组,那么实际有效的Si-Si原子对数量为 (c-a)/2, 无效的键级数据有 a + (c-a)/2 = (c+a)/2,其中 a 组数据是原子和原子本身的键级,
如,Si1-Si1,Si2-Si2,...,另外 (c-a)/2 是被重复计算的数据,如 Si1-Si2 和 Si2-Si1 应当只保留其中之一,故实际有效
的Si-Si原子对数量为 (c-a)/2。
3. 运行上述代码需要将 40_Mayer键级与键长_升级版.py 计算出来的同种原子对键级数据保存到一个新的txt文件中,不包含表头,参考数据可格式如下
1:1 0.0 4.153
1:5 2.76 0.598
1:164 2.508 0.753
1:182 2.371 0.873
1:213 2.395 0.853
......
4. 脚本清洗无效和重复数据的原理。通过分析上述第一列数据中原子对的编号是否相同,以及原子对互换后是否与其他数据重复来实现数据清洗。
5. 两个脚本保存数据的文件名依次为 bondOrder_retain.txt 和 bondOrder_retain_2nd.txt。
注意:该脚本是在40_Mayer键级多中心原子综合版.py基础上进一步开发的
1. 升级版除了能够计算键级外,还能够计算相应原子对的键长,从而绘制键长和键级的变化关系图。
2. 计算完后原子对,键长以及键级数据会以追加的形式写入到bondOrder.txt文件,复制到origin进行绘图即可,数据的结构如下所示,共有3列,分别为原子对编号,键长和键级。
atomPair bondLength bondOrder
1:1 0.0 4.153
1:5 2.76 0.598
1:164 2.508 0.753
1:182 2.371 0.873
1:213 2.395 0.853
......
3. 该脚本计算非同类原子键级时,如Si-O,输出的数据无需清洗,可直接用于绘图;如果计算的是同类原子键级,输出的键级数据中会有部分重复数据,需要基于 40_同种原子键级计算.py
脚本 或者 40_同种原子键级计算_2nd.py 脚本进行清洗。
注意本脚本是基于multiwfn输出的bndmat.txt文件进行计算的
1. 首先提示用户输入xyz轨迹文件明,输入用于电子结构计算的xyz文件名即可
2. 提示输入模拟盒子尺寸,默认是正方形盒子,用于考虑周期性边界
3. 提示输入中心原子和配位原子
4. 提示输入原子对的截断半径
5. 提示输入含有键级数据的矩阵文件名,默认是multiwfn输出的bndmat.txt文件
6. 最后输出键级数据文件bondOrder.txt,包含每一个中心原子及其配位原子的键级数据,平均值,方差,截断半径等。
注意:该脚本仅能计算 中心原子和配位原子 不是同类原子的键级数据,如Si-O原子对,对于同类原子对的键级,如Si-Si原子对,则需要采用 40_Mayer键级与键长_升级版.py 脚本来计算。
- 周期性边界条件以及周期性扩增的原理示意图如下:
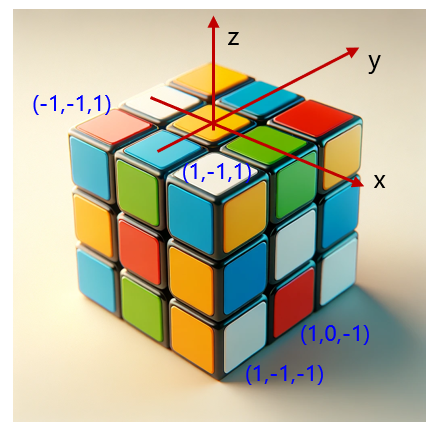
假设盒子的晶格常数分别为a,b,c,如果需要将盒子在x,y和z的正方向(1,1,1)方向扩增一倍,则相应坐标变换为 (x+a,y+b,z+c);如果是负方向,则是(-1,-1,-1),相应坐标变换是(x-a,y-b,z-c);如果在三维空间的所有方向都扩增一倍,即有 9*3-1=26种组合,其中(0,0,0)代表原始盒子坐标。
请选择单质数据库,1 2, or 3?
1= pureDensityGasDict (单质密度数据库,适用于合金熔体体系,暂不能计算含有H,N,O,F,Cl,Ar等元素的混合体系密度),
2= pureDensitySolidDict (单质密度数据库,适用于熔体体系中含有H,N,O,F,Cl,Ar等非金属元素体系),非金属元素的密度基于氧化物密度倒推校正获得,暂不完善
3= pureSolidOxideDensityDict (纯固态氧化物密度数据库),参考手册 The Oxide Handbook,HRC等
4= pureLiquidOxideDensityFactsageDict (纯液态氧化物密度数据库), 源于factsage倒退修正,1800k
5= pureOxideDensityMSDict (纯固态氧化物密度数据库),参考 Material Project晶体结构数据库
6= mixOxideSimpleSubstanceDensityDict (金属和氧化物混合体系密度数据库,适合两相混合体系计算,参考1和4),推荐使用该数据库
合金混合体系相对分子质量计算和Factsage密度计算可以任选1和2数据库
🟢 升级版本参考:49_[扩展Beta]单质或化合物或多相混合体系密度计算.py
1. 将 Materianl Studio 建模导出的.cell文件转换为同名的.xyz文件,并统计各原子的数量,保留晶胞长度信息。
2. 可视化软件Ovito能实现类似的格式转换
该脚本的缺点在于未考虑盒子的周期性
以下是代码的功能分条总结:
1. 从文件中读取坐标数据并将其绘制为散点图。
2. 提示用户选择文件路径或手动输入文件名。
3. 读取文件并计算原子数。
4. 创建原子数原子数的多维矩阵。
5. 从文件中读取键级数据并将其填充到矩阵中。
6. 提取用户指定的中心原子和配位原子的键级数据。
7. 计算键级数据的平均值。
8. 将键级数据和平均值写入文件。
该代码主要实现了以下功能:
1. 导入了时间模块,并显示了当前时间。
2. 提示用户输入三个点的坐标和晶格常数a,并将输入的字符串转换为浮点数。
3. 根据输入的三个点的坐标,计算出平面方程的系数A、B、C、D,并显示方程形式和系数。
4. 根据平面方程的系数,计算出x、y、z轴截距,进而计算出晶面指数h、k、l,并显示晶面指数。
5. 根据晶面指数和晶格常数a,计算出立方晶系晶面间距d,并显示结果。
6. 根据平面方程的系数,计算出原点到平面的距离r,并显示结果。
7. 计算r/d的值,并显示结果。
8. 显示任务完成,并计算程序运行时间。
-
功能:推荐运行在超算,遍历当前目录下的
tem.out文件(cp2k输出文件),查找包含“SCF run converged in”的行提取SCF步数以及包含“MD| Step number”的行提取MD步数。然后将这两组数据配对写入step_scf.txt文件中,实现将每个MD步对应的SCF收敛步数输出到文件的功能。 -
生成的
step_scf.txt文件,可以使用本地 01_plot_colvar.py 脚本进行绘图。 -
编程思路:
能不能编写一个bash脚本实现以下功能:
1. 当前目录下有一个 tem.out 文件,里面有很多行,现在我需要提取类似如下两行中的数据,并且把它写入到 step_scf.txt 文件中,分为两列,第一列数据是 "MD| Step number",第二列数据是 "SCF run converged in" ,写入到的txt文件不需要表头,两列数据使用空格分隔。
2. tem.out中相关行示例:
*** SCF run converged in 8 steps ***
MD| Step number 4203
注意:SCF run converged in在tem.out文件中会先出现,然后再出现对应的 MD| Step number 行。
3. 建议先遍历 tem.out文件,提取 "MD| Step number" 行的数值作为第一列,然后再遍历文件,提取 "SCF run converged in" 行的数值作为第二列。
4. 获取当前命令执行的所在目录,将 step_scf.txt 文件保存到当前目录下。
- 相关命令行alias
# 通过命令行查看 tem.out 文件中的相关步数
alias step='grep "MD| Step number" tem.out'
alias scfp='grep "SCF run converged in" tem.out | nl'
# 将该脚本放在超算服务器下的某一路径,通过 sfmd 在命令执行路径下生成 step_scf.txt 文件,不需要在每一个 tem.out 路径下放置一个 21_extract_scf_step.sh 脚本
alias sfmd='bash /public21/home/sc90511/tool_user_defined/21_extract_scf_step.sh'- 结果示例
-
功能:读取CP2K输出的ener文件,提取离子步、时间和耗时等数据,并根据用户选择绘制离子步耗时的变化曲线。用户可选择绘制所有步数或分段绘制,程序会计算并输出平均耗时,同时支持自定义刻度和保存图片。
-
结果示例
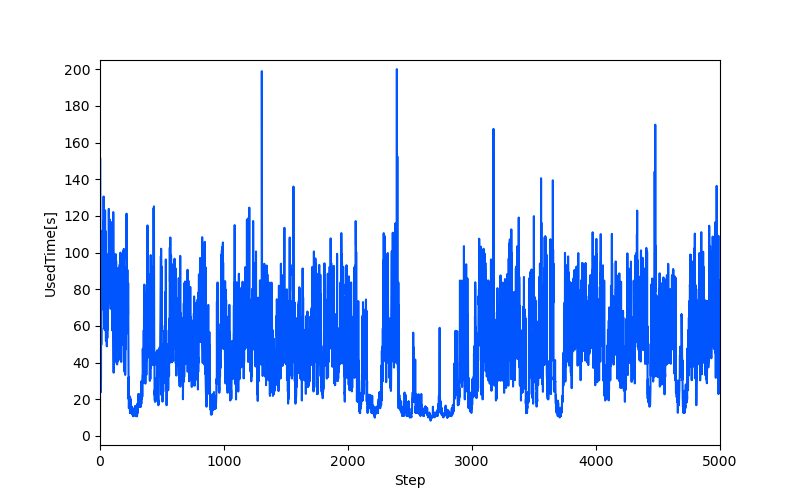
-
功能:从CP2K输出的ener文件中提取步数、时间、温度和势能等数据,并根据用户指定的步数分界来选择数据区间。随后,它利用Matplotlib绘制了温度与步数和势能与时间的变化图,同时提供了保存图像的选项。
-
20_ener分步绘温度能量图.py相比于12_ener文件绘温度能量图.py脚本,增加了数据区间划分的功能,使用户可以根据指定的分界步数选择数据子区间进行绘图。
注意:该功能已经耦合到 58_[47类]轨迹分析和绘图.py 脚本的 05方法 中。
- 结果示例
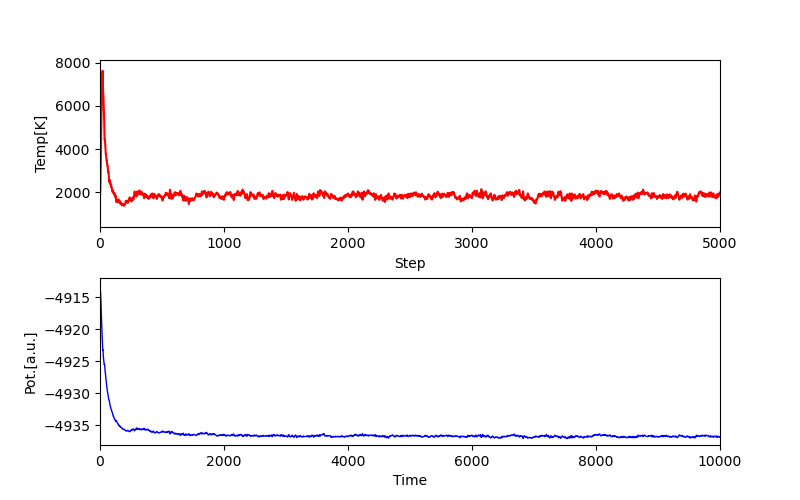
-
功能:在多帧xyz文件中,根据用户指定的原子编号和截断半径,筛选出每一帧中距离指定原子较近的其他原子,并将每帧的筛选结果(包括统计数量和具体坐标信息)写入到一个新的xyz格式文件中。
注意:19_[超快速] 特定编号原子周围半径r范围内原子.py相比于19_特定编号原子周围半径r范围内原子.py,唯一的区别在于注释了很多 print 语句。
-
功能:对cp2k输出的ener文件数据的整体处理与绘图。程序会读取文件中除首行标题外的所有数据,并绘制两幅图:上图显示温度随步数变化的曲线,下图显示势能随时间变化的曲线。
对于xyz轨迹文件,输入想要提取的起始帧数和截止帧数来提取某一帧或者连续的几帧,相关升级功能已经耦合到 58_[47类]轨迹分析和绘图.py 的功能04中